Quick start
Analyses with shinyseg are easy and highly flexible. All you need to do is upload a pedigree file, enter the genetic/clinical data, and specify a suitable penetrance model. The following sections briefly explain how to do so with an example case, but you can find detailed guidance within the app or in the documentation.
Uploading a pedigree
The application works with pedigree files in ped format, like those created by QuickPed. Here is an example of such a file and the corresponding family:
id fid mid sex
1 0 0 1
2 0 0 2
3 1 2 1
4 0 0 1
5 1 2 2
6 4 5 1
7 4 5 1
![]()
Note that other columns may be present, but you will at least need: id
(individual ID), fid (father’s ID, 0 if not included in the pedigree),
mid (mother’s ID, 0 if not included in the pedigree), and sex (1 =
male; 2 = female).
You can even upload multiple families at the same time. Read here how to do it!
Genetic and clinical data
Once a pedigree is loaded, users are presented with a table to enter the analysis data:
- phenotype: free-text field to specify disease phenotypes
relevant to the analysis, e.g.
affected,breast cancer, etc.nonafffor unaffected individuals, empty if unknown. - carrier:
negfor non-carriers,hetfor heterozygous (or hemizygous) carriers,homfor homozygous carriers. Empty if unknown. - proband: checkbox indicating the proband or index case.
- age: integer between 1-100 specifying the age of disease onset or censoring.
For instance:
id fid mid sex phenotype carrier proband age
1 0 0 1 . . . 80
2 0 0 2 . . . 80
3 1 2 1 affected het . 40
4 0 0 1 nonaff neg . 60
5 1 2 2 nonaff het . 60
6 4 5 1 affected het 1 40
7 4 5 1 affected het . 40
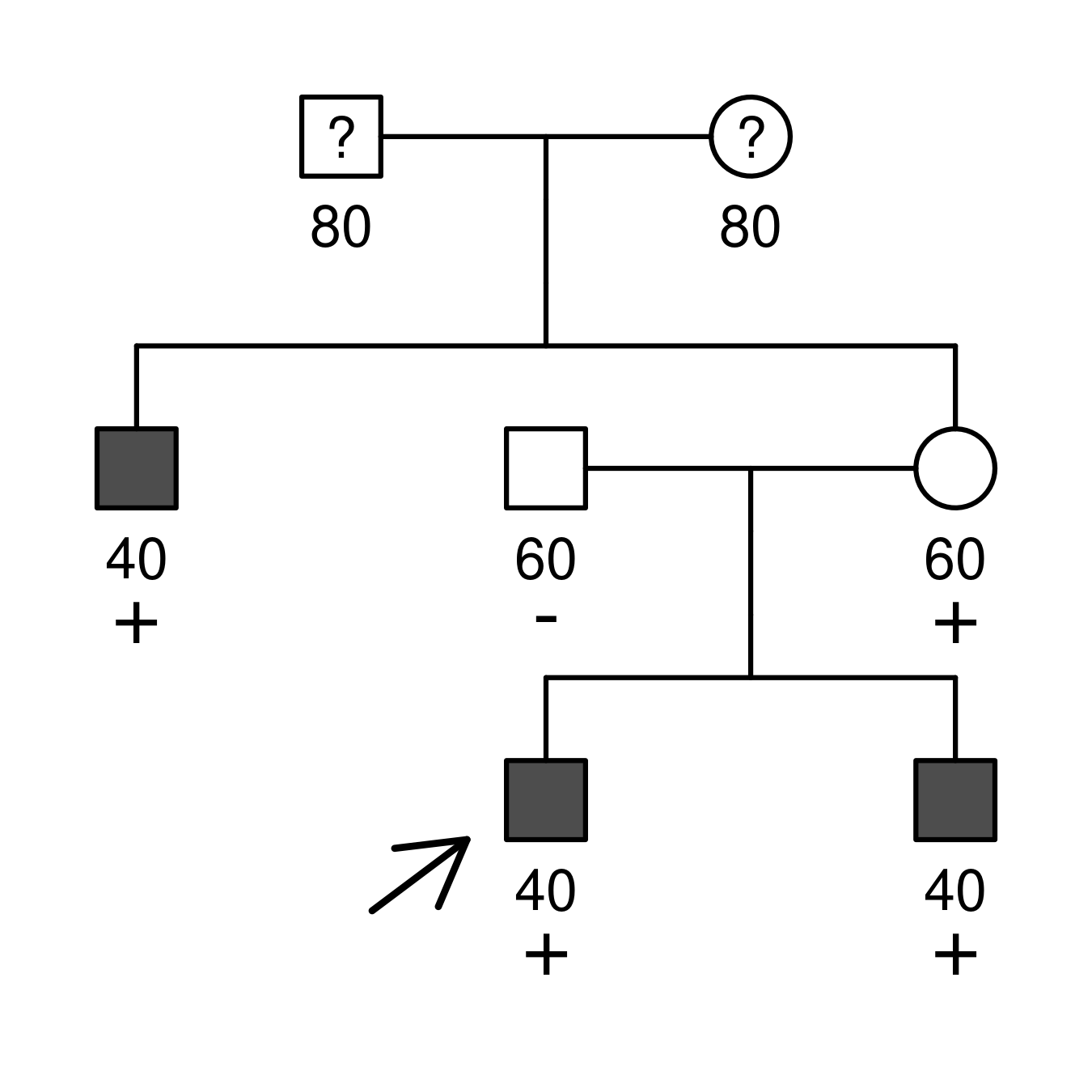
The app will signal if some information is wrong. Look at the requirements here!
Penetrance model
The inheritance pattern combines choices for chromosome (A: autosomal,
X: X-linked) and dominance (D: dominant, R: recessive, I:
incomplete dominance). In this example we select X-linked recessive
(XR), which will adjust other inputs accordingly.
Next, it is time to assign the probabilities of observing the analysis phenotypes conditional on carriership status. These will be hereafter referred to as penetrances, and their specification may be done in two ways:
1) Relative risk
A parametric version of the survival penetrances described in Belman et al. (2020). It is based on:
- Baseline lifetime risk, mean and SD: the lifetime risk, mean, and standard deviation of disease onset in non-carriers and heterozygous carriers in recessive inheritance.
- Hazard ratios: the relative risks in homo-, hemi-, and heterozygous carriers in dominant inheritance, compared to the baseline. They can be either constant or age-dependent and may also be specified through a variant-associated lifetime risk.
For example, the following describes a constant relative risk model in
which heterozygous women (♀het) and non-carriers (neg) have a
lifetime risk (phenocopy rate) of 1% with onset at 70±15 years of age,
while hemizygous men (♂het) and homozygous women (hom) have a
lifetime risk of 75%.
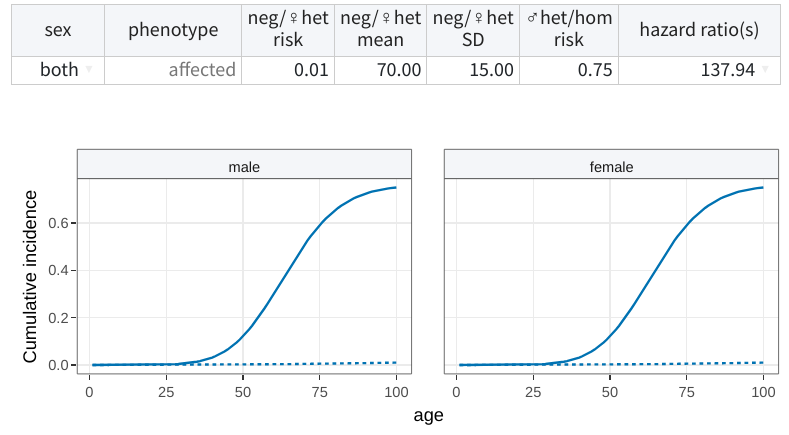
To facilitate the specification, shinyseg can also optimize these parameters based on user-provided cumulative incidence data. See here how to do it!
2) Liability class
Alternatively, users can manually define the penetrances using a table. The following represents the simplest case where there are no phenocopies, and the chance of disease onset in hemizygous men and homozygous women is 100%.

More detailed specifications, dependent on sex, phenotype, and age, can be created by adding more rows and filling in these columns accordingly.
Cosegregation evidence
Calculating the FLB is then as easy as clicking a button. For instance,
with a population variant frequency of 0.001 and the simple liability
class model from before, we get an FLB = 8.00. shinyseg reports this
as supporting evidence for pathogenicity based on Jarvik and Browning’s
(2016) thresholds.

Afterward, the app opens up more possibilities, including performing sensitivity analyses to assess the robustness of your results.
Take a look at the documentation to explore this feature!